
Phage-Bacteria Interactions During Disease
The waterborne pathogen Vibrio cholerae is the etiological agent of cholera, an infectious diarrheal disease that sickens and kills only those already burdened by poverty, conflict, or natural disasters. In both the aquatic environment and the human gut, V. cholerae is attacked by predatory phages (vibriophages). Interestingly, cholera patients can shed both vibriophages and viable V. cholerae, indicating that complex interactions between phages and their hosts take place within the human intestine, as neither entity is driven to extinction. The ability of V. cholerae to prevent phage predation is critical for its evolutionary fitness and epidemic potential. In turn, as obligate bacterial parasites, phages must co-evolve to overcome this resistance or they will face extinction. Our research is aimed at understanding the bacterial immunity and opposing phage immune evasion strategies at play in this dynamic co-evolutionary arms race. We use a combination of genomic and mechanism-driven approaches to identify and experimentally validate such strategies in disease associated phage and V. cholerae isolates.
ICP1 is a lytic bacteriophage of V. cholerae. When V. cholerae encodes defenses against ICP1, ICP1 infections are not productive, no ICP1 progeny are made, and V. cholerae survives. However, when ICP1 is equipped with counter-defenses, ICP1 infections are productive, many ICP1 progeny are made and V. cholerae dies.
Our Approach: Following V. cholerae - Phage Conflict in a Cholera Endemic Region
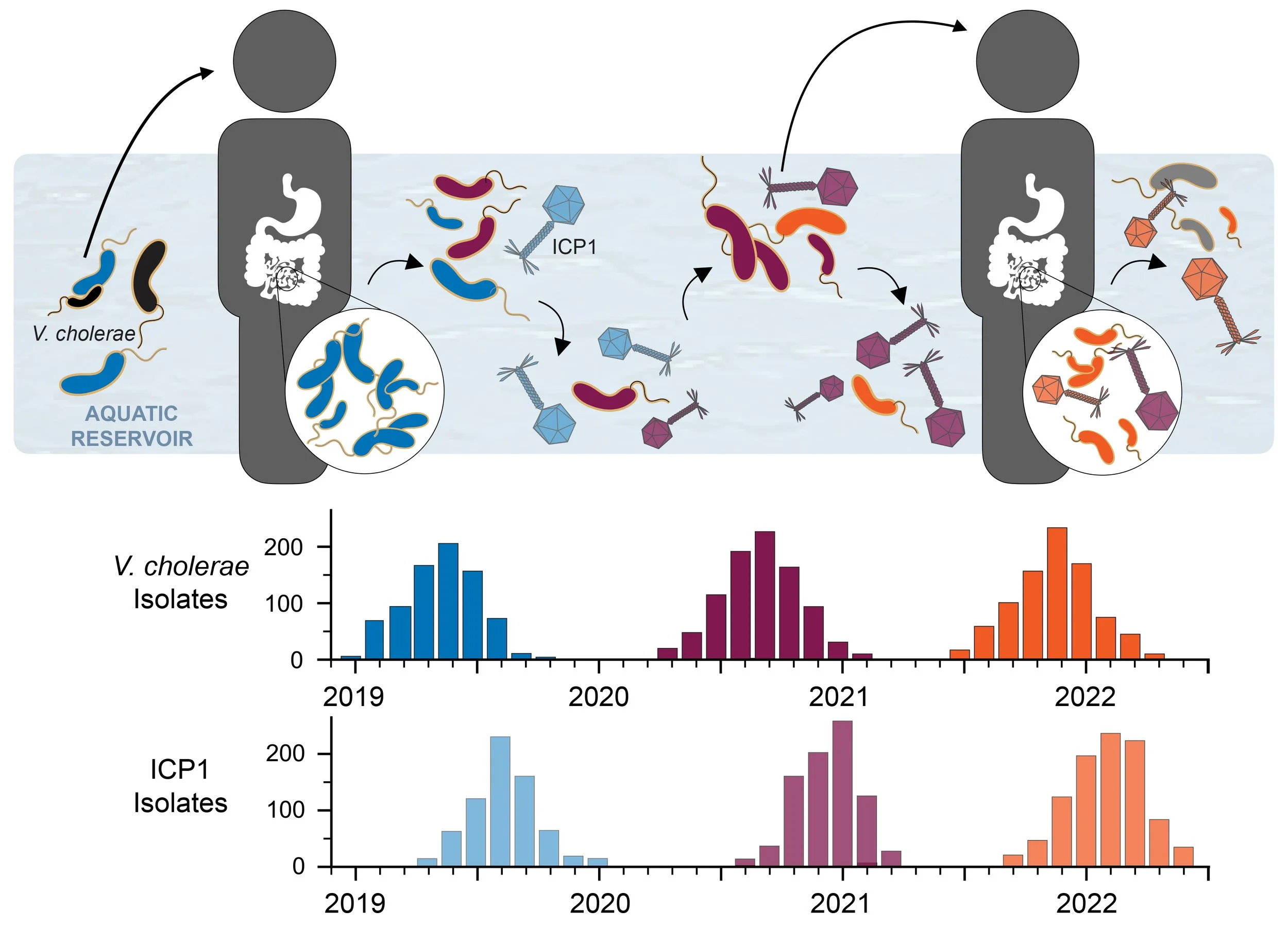
Clinical surveillance reveals the arms race between V. cholerae and ICP1.
In collaboration with the icddr,b (International Center for Diarrheal Disease Research, Bangladesh), we isolate V. cholerae and phages (mainly ICP1) from cholera patient stool samples. Our work has revealed a continuing arms race between V. cholerae and ICP1.
We see cyclical patterns of positive selection for phage defense in V. cholerae, followed by positive selection of ICP1 with appropriate counter-defenses. As schematized above, an abundant V. cholerae strain (blue) encodes resistance to previously circulating ICP1 strains. ICP1 then evolves a counter-defense (light blue), allowing it to infect these abundant V. cholerae strains. In response, V. cholerae acquires a new defense (maroon) against the now prominent ICP1 and the bacterial population blooms. Acquisition of a novel counter-defense (light maroon) allows ICP1 to infect and kill these abundant V. cholerae and decrease the population size. The cycle continues (orange). Our work has identified many of the molecular mechanisms driving these patterns.
More details of these defenses and counter-defenses described by our lab can be found below.
Novel mobile genetic elements that mediate defense against phages, and how ICP1 fights back
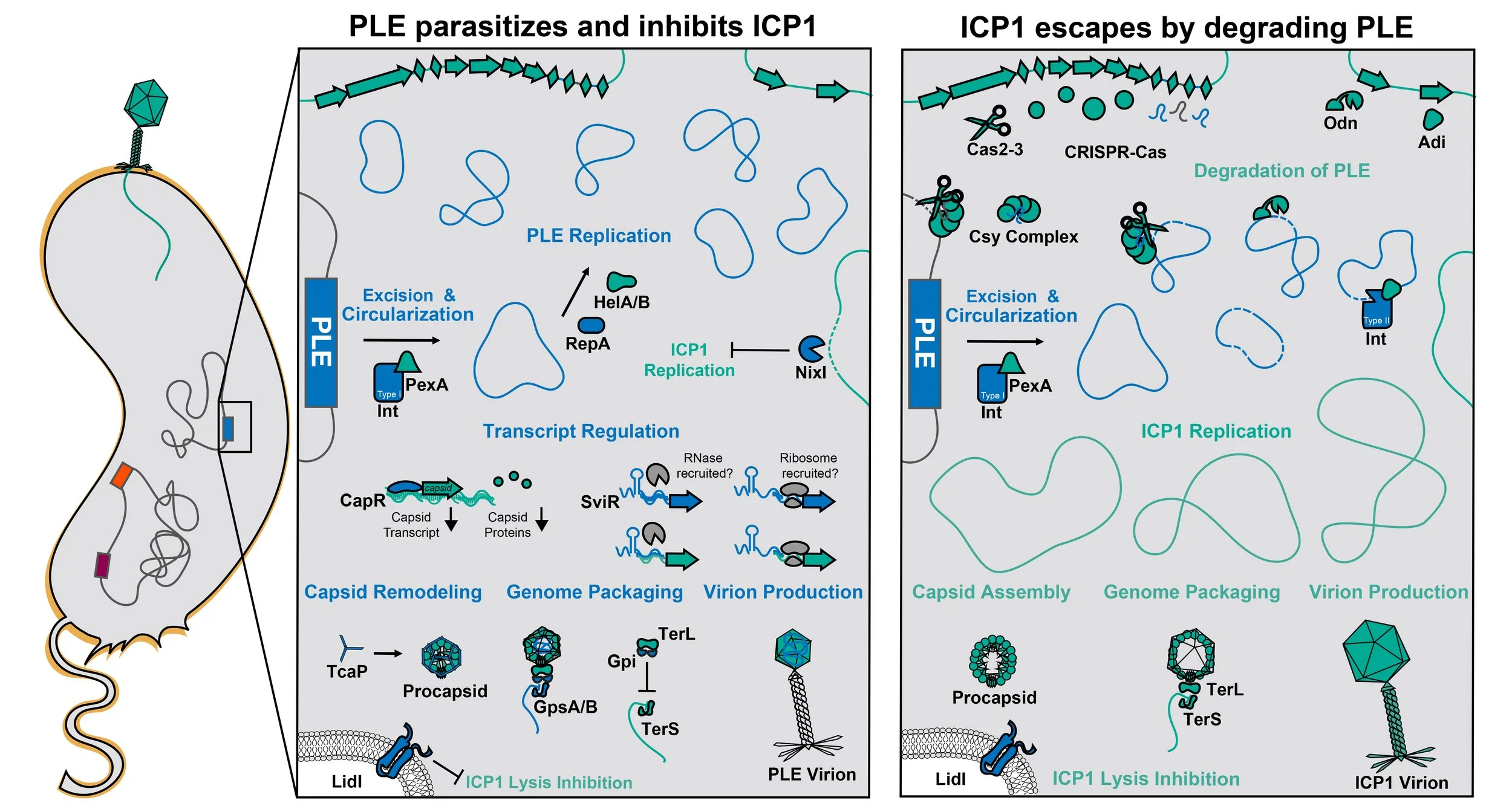
We discovered a family of dynamic anti-phage MGEs called phage-inducible chromosomal island-like elements (PLEs) as an enduring feature of V. cholerae’s genome that provide robust and specific protection against ICP1. We established that the PLE defense strategy is based on hyperparasitism, whereby PLEs abolish ICP1 production while exploiting phage resources for their own selfish spread, ultimately benefiting the V. cholerae host population.
A primary goal of our research has been to identify the mechanisms underlying PLE activity. We characterized PLEs’ response to phage infection and how PLE perturbs ICP1’s lifecycle using systems-level -omics approaches and complementary mechanism-driven studies.
Following infection by ICP1, PLE uses PLE- and ICP1-encoded proteins (blue and green, respectively) to excise from the V. cholerae chromosome and replicate (RepA, HelA/B). PLE deploys the nickase, NixI, to limit the phage’s replication. PLE manipulates phage gene expression using a transcriptional regulator (CapR) and a regulatory small RNA (SviR). To hijack and manipulate the structural proteins made by ICP1 that PLE needs for its own virion assembly, PLE remodels capsids using the external scaffolding protein (TcaP). PLE encodes packaging specifiers (GpsA/B) to redirect the phage’s packaging motor (TerL) to package PLE genomes and a packaging inhibitor (Gpi) to block ICP1’s genome from being packaged. Ultimately, PLE assembles virions carrying the replicated PLE genomes that are released from the infected cell with the help of a membrane protein (LidI). Given the many redundant pathways that PLE uses to inhibit ICP1, PLE completely blocks the production of ICP1 progeny, thus protecting neighboring cells from phage infection.
Our surveillance efforts have shown that ICP1 survives in the face of the highly inhibitory PLE by evolving counter-defenses that target and degrade the PLE genome. To date, three nuclease-effectors against PLE have been identified in ICP1 isolates, 1) CRISPR-Cas, 2) Odn, and 3) Adi. CRISPR-Cas carries spacers against the PLE genome that guide the nuclease effector (Cas2-3) to target the PLE genome for degradation. Odn has a DNA-binding domain with specificity for PLE’s origin of replication and a nuclease domain that degrades the PLE genome. Adi specifically targets a subset of PLEs (those with Int Type II) and, through currently unknown mechanisms, triggers nucleolytic cleavage of the PLE genome. Without a template to replicate or express its inhibitors of ICP1, PLE loses the capacity to block the amplification of ICP1. ICP1’s lifecycle carries on as it would in the absence of PLE, wherein ICP1 replicates its genome, assembles capsids and tails (not pictured), packages its genome, and releases ICP1 virions. When many ICP1 virions are present in the surrounding environment, ICP1 exhibits lysis inhibition, stalling lysis of infected cells to allow for the assembly of more virions prior to lysis.
Current projects in the lab are focused on understanding how PLE is transcriptionally activated upon phage infection and deciphering the mechanisms of additional PLE-encoded factors that block ICP1 production. We also want to understand the molecular mechanisms underpinning the selection and dynamics of emergent PLEs.
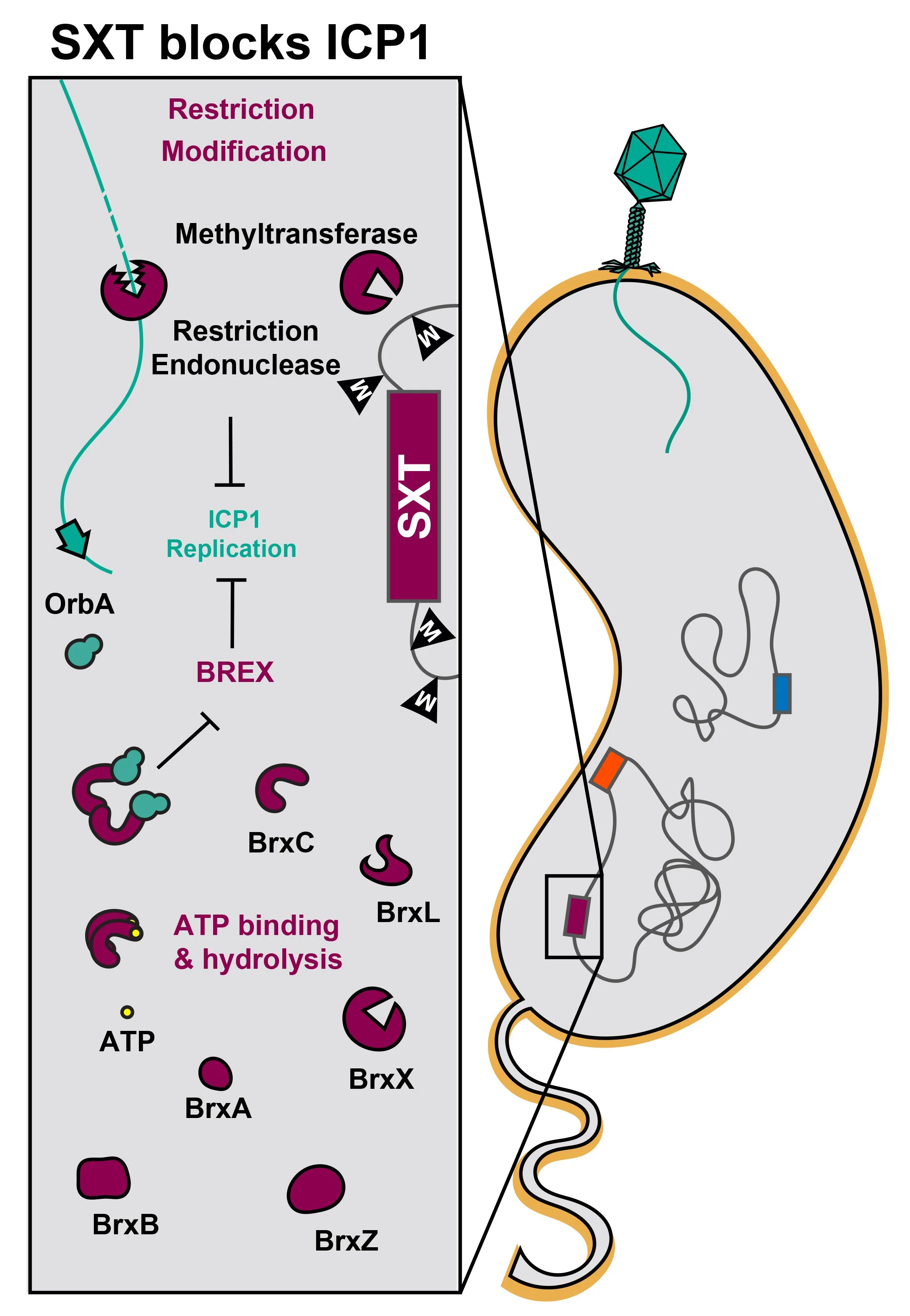
SXT Integrative and Conjugative Elements (ICEs) (maroon), which are notorious vectors of antibiotic-resistance genes, also invariably encode phage defense systems. Two variants of SXT ICEs most commonly found in recent V. cholerae clinical isolates from Bangladesh encode either Restriction Modification (RM) systems or the bacteriophage exclusion (BREX) system. For RM systems, the addition of methyl groups to DNA provides protection from the restriction endonuclease. ICP1 phages (teal) that have not been modified at the restriction sites are thus sensitive to the RM system, and their genomes are degraded during infection. For the BREX system, the mechanism of restriction is unknown and an active area of study in our lab. ICP1 isolates with OrbA can escape BREX restriction through a direct interaction between OrbA and BrxC.
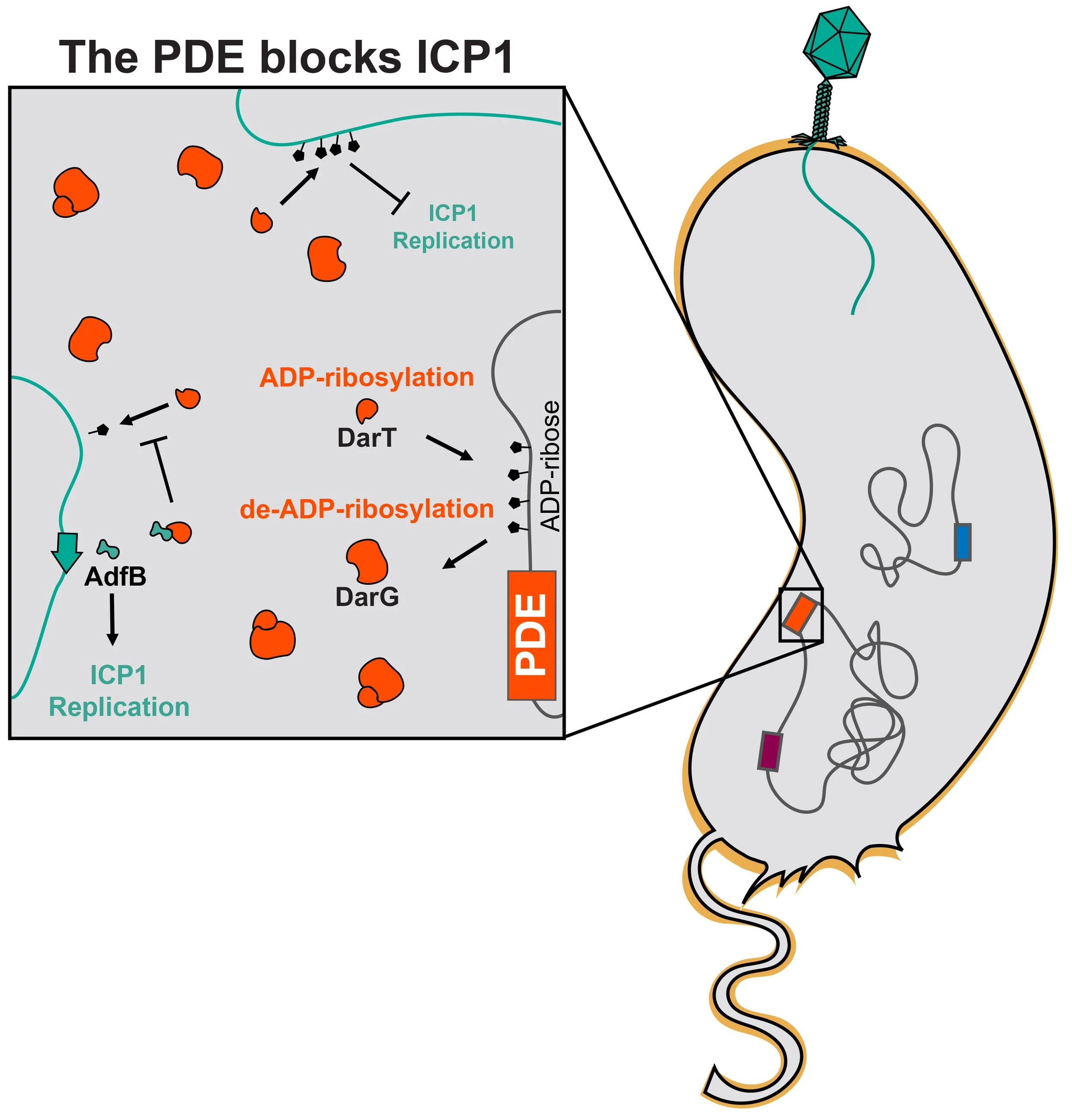
The Phage Defensive Element (PDE) (orange) encodes a toxin-antitoxin system, DarTG. The toxin, DarT, adds ADP-ribosyl groups to DNA and the antitoxin, DarG, catalyzes the removal of these ADP-ribosyl groups, ultimately maintaining an equilibrium that allows for cell survival. During phage infection, this equilibrium is disrupted, resulting in excessive ADP-ribosylation. ADP-ribosylation of the incoming ICP1 genome (colored teal) blocks phage replication, resulting in no ICP1 progeny production. In this case, phage infection is not productive, but the infected cell still dies, ultimately sparing the rest of the population from phage attack. Some ICP1 isolates encode a counter-defense, AdfB, which blocks DarT’s activity and allows for ICP1 progeny production in the presence of the DarTG system. We want to understand how DarTG is activated upon phage infection.